
Profile
Professor Aleksandra Filipovska received her PhD in 2002 from the University of Otago, New Zealand. From 2003-2005 she was a NZ Foundation for Research, Science and Technology Fellow at the MRC Mitochondrial Biology Unit in Cambridge, the United Kingdom. In 2006 she relocated to Australia as a NHMRC Howard Florey Fellow and established her Mitochondrial Medicine and Biology research group at the Harry Perkins Institute for Medical Research. She was an Australian Research Council Future Fellow from 2009 to 2014 and currently she is a NHMRC Senior Research Fellow and Research Professor at The University of Western Australia and Deputy Director ARC Centre of Excellence in Synthetic Biology. Her research interests are in the regulation of mitochondrial gene expression by RNA-binding proteins in health and disease. In addition her research group uses next generation technologies to identify pathogenic mutations in mitochondrial genes that cause mitochondrial disease in genetically isolated populations.
Research overview
Mitochondria are microscopic, energy producing machines that are found in virtually all eukaryotic cells. Mitochondria are essential for the normal function and survival of all eukaryotic organisms. Mitochondria produce more than 90% of the energy required by our bodies and thereby have a fundamental role in cell and energy metabolism. Mitochondria are composed of proteins encoded by both the nuclear and mitochondrial genome and the coordinated expression of both genomes is essential for energy production. Impaired energy production leads to mitochondrial dysfunction that causes or contributes significantly to a variety of diseases including metabolic disorders, neurodegenerative and cardiovascular diseases, diabetes, obesity, cancer and ageing. Mitochondrial dysfunction is caused by mutations or variations in nuclear or mitochondrial genes that encode proteins or regulatory RNAs essential for mitochondrial biogenesis. How uncoordinated gene expression causes mitochondrial dysfunction and compromised energy production in common diseases is poorly understood, making it difficult to develop effective treatments. To unravel how mitochondrial function fails and to identify therapeutic targets in our group we seek to understand how gene expression is regulated between mitochondria and the nucleus and how this regulation is disrupted in disease. We use synthetic biology and genome editing tools to create unique and physiologically relevant models of human diseases caused by impaired gene expression. We use our models to identify how changes in the mitochondrial and nuclear genomes cause mitochondrial dysfunction in neurological, cardiovascular and metabolic disease, in cancer and in ageing. We identify targets and develop drugs towards them to provide potential treatments for these diseases and improve energy function.
Mitochondrial biogenesis is intimately dependent on the coordinated expression of the nuclear and mitochondrial genomes that is necessary for the assembly and function of the respiratory complexes to produce most of the energy required by cells. Although highly compacted in animals, the mitochondrial genome and its expression are essential for survival, development and optimal energy production. The machinery that regulates gene expression within mitochondria is localized within the same compartment and, like in their ancestors, the bacteria, this machinery does not use membrane-based compartmentalization to order the gene expression pathway. Therefore, the lifecycle of mitochondrial RNAs from transcription through processing, maturation, protein synthesis to turnover is mediated by a gamut of RNA-binding proteins, all contained within the mitochondrial matrix milieu. Recent discoveries indicate that multiple processes regulating RNA metabolism occur at once but since mitochondria have a new complement of RNA-binding proteins, many evolved de novo from nuclear genes, we are left wondering how coordinated are these processes? In our group we investigate the coordinated and stochastic processes that govern the mitochondrial transcriptome. Discoveries from the last decade have revealed the complexity of mitochondrial gene expression and the need for its in-depth exploration to understand how these organelles can respond to the different energy demands of cells.
Image: Delivery of nanoparticles into tumors to promote gene silencing and sensitize cancer cells to chemotherapy
Research projects
1. Mitochondrial RNA-binding proteins and their role in mitochondrial gene expression
Mitochondria play a fundamental role in cell and energy metabolism and consequently mitochondrial dysfunction can lead to severe multi-system disorders with wide range of clinical presentations that commonly include neurodegeneration, muscle defects and exercise intolerance. To understand these conditions better and identify therapeutic targets it is necessary to understand how gene expression is regulated within mitochondria, as some of the most significant gaps in our knowledge of mitochondrial function and disease are in the regulation of mitochondrial gene expression. Links between transcription and translation in mammalian mitochondria are not well understood.
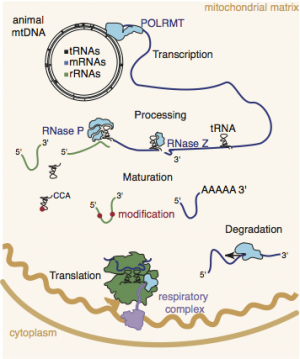
Mitochondrial mRNAs are transcribed as part of long primary transcripts that generally encompass the entire mtDNA, therefore the ratios of the 11 mammalian mitochondrial mRNAs and their proteins are controlled post-transcriptionally.
Little is known about how these 11 mRNAs are regulated in mammalian mitochondria. This is particularly important since tissue-, cell- and disease-specific variations in expression of mitochondrial RNAs has been observed, but cannot be explained at present. The basic components and mechanisms of transcription have recently been discovered, however the control of mRNA processing, translation and stability remains unclear.
2. Identification of mutations that cause mitochondrial disease
Mitochondrial diseases are progressive and debilitating multi-system disorders that occur as a result of mutations in nuclear or mitochondrial genes with no known cures to date. The clinical heterogeneity in mitochondrial disorders is complemented by genetic heterogeneity, where mutations in mitochondrial or nuclear genes cause similar phenotypes thus complicating mutation identification. We use next generation technologies to identify mutations in DNA from patients that suffer from mitochondrial diseases to identify the mutations that cause these diseases. We use patient cells to investigate how mutations in mitochondrial genes cause the molecular changes that cause mitochondrial and cellular dysfunction that leads to the disease pathology.
3. Characterizing the pathology of mitochondrial diseases
Mitochondrial diseases are progressive and debilitating multi-system disorders that occur as a result of mutations in nuclear or mitochondrial genes at a frequency of up to 1 in 4,000 live births with no known cure. Mutations in nuclear genes that code for mitochondrial proteins have been found to cause a range of diseases including mitochondrial diseases that have the same pathologies to those observed in patients with mutations in mtDNA. Our group works towards developing several animal models of mitochondrial disease and we investigate the effects of mitochondrial dysfunction in different tissues. Furthermore we are investigating how specific proteins regulate gene expression and how lack of these genes can cause the disease pathology. Our goal is to understand the molecular mechanisms underlying mitochondrial disease and provide new avenues for therapeutic interventions. Furthermore we are developing treatments for our established models of mitochondrial disease by synthesising specific small molecule drugs or repurposing of already available drugs.
4. Development of new technologies to investigate mitochondrial gene expression
We are developing new next generation technologies and analytical methods to investigate mitochondrial transcripts, their processing, translation and the role of mitochondrial RNA-binding proteins in cell and mouse models of disease. Furthermore we create new assays and arrays to identify the RNA targets of mitochondrial RNA-binding proteins and modulate mitochondrial gene expression.
Selected Publications
1. Liu, G., Mercer, T.R., Shearwood, A.-M.J., Siira, S.J., Hibbs, M.E., Mattick, J.S., Rackham, O. and Filipovska, A. (2013) Mapping of mitochondrial RNA-protein interactions by digital RNase footprinting. Cell Reports 5(3):839-48. [NCBI PubMed Entry]
2. Richman, T.R., Ermer, J.A., Davies, S.M., Perks, K.L., Viola, H.M., Shearwood, A.-M.J., Hool, L.C., Rackham, O. and Filipovska, A. (2015) Mutation in MRPS34 compromises protein synthesis and causes mitochondrial dysfunction. PLOS Genetics10.1371/journal.pgen.1005089. [NCBI PubMed Entry]
3. Small, I.D., Rackham O. and Filipovska, A. (2013) Organelle transcriptomes: innovations on a prokaryotic blueprint. Current Opinions in Microbiology 16(5):652-8. [NCBI PubMed Entry]
4. Coquille, S. *, Filipovska, A.*, Chia, T., Rajappa, L., Lingford, J.P., Razif, M.F., Thore, S. and Rackham, O. (2015) An artificial PPR scaffold for programmable RNA recognition. Nature Communications 5:5729. doi: 10.1038/ncomms6729. [NCBI PubMed Entry] *co-first authors
5. Richman, T.R., Davies, S.M.K., Shearwood, A-M.J., Ermer, J.A., Scott, L.H., Hibbs, M.E., Rackham, O. and Filipovska, A. (2014) A bifunctional protein regulates mitochondrial protein synthesis. Nucleic Acids Research 42(9): 5483-94. [NCBI PubMed Entry]
6. Rackham, O., Shearwood, A.-M.J., Mercer, T.R., Davies, S.M.K., Mattick, J.S. and Filipovska, A. (2011) Long non-coding RNAs are generated from the mitochondrial genome and regulated by nuclear-encoded proteins. RNA 17(12): 2085-93. [NCBI PubMed Entry]
7. Mercer, T.R., Neph, S., Crawford, J., Dinger, M.E., Smith, M.A., Shearwood, A.-M.J., Haugen, E., Bracken, C.P., Rackham, O., Stamatoyannopoulos, J.A., Filipovska, A.* and Mattick, J.S. (2011) The human mitochondrial transcriptome. Cell 146(4): 645-658. [NCBI PubMed Entry] *co-corresponding author
8. Filipovska A., Razif M.F.M., Nygård K.K.A. and Rackham O. (2011) A universal code for RNA recognition by PUF proteins. Nature Chemical Biology 7(7): 425-7. [NCBI PubMed Entry]
9. Lopez Sanchez, M.I.G., Mercer, T.R., Davies, S.M., Shearwood, A.-M.J., Nygård, K.K.A., Richman, T.R., Mattick, J.S., Rackham, O. and Filipovska, A. (2011) RNA processing in human mitochondria. Cell Cycle 10(17): 1-13. [NCBI PubMed Entry]
10. Rackham O., Davies, S.M.K., Shearwood, A.-M.J., Hamilton, K.L., Whelan, J. and Filipovska, A. Pentatricopeptide repeat domain protein 1 lowers the levels of mitochondrial leucine tRNAs in cells (2009) Nucleic Acids Research 37(17):5859-67. [NCBI PubMed Entry]